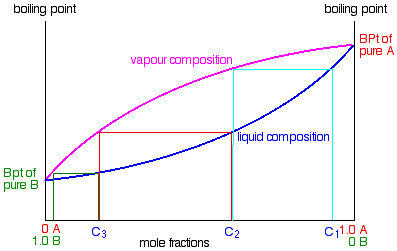
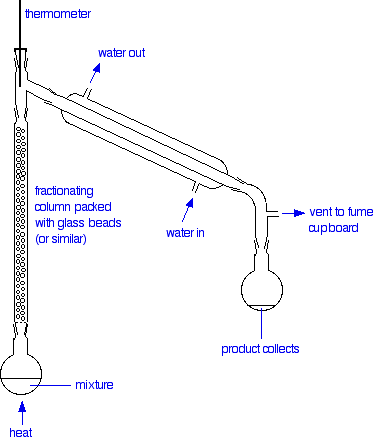
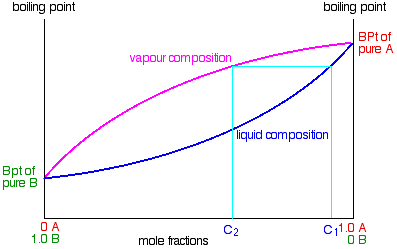
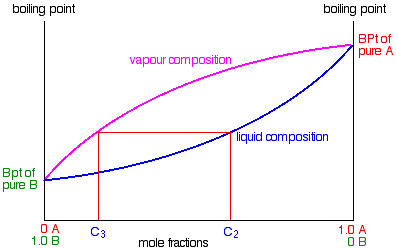
So what happens to that liquid now? It will start to trickle
down the column where it will meet new hot vapour rising. That will cause the already condensed vapour to reboil.
Some of the liquid of composition C2 will boil
to give a vapour of composition C3. Let's concentrate first on that new vapour and think about the unvaporised
part of the liquid afterwards.
The vapour
This new vapour will again move further up the fractionating
column until it gets to a temperature where it can condense. Then the whole process repeats itself.
Each time the vapour condenses to a liquid, this liquid will
start to trickle back down the column where it will be reboiled by up-coming hot vapour. Each time this happens the new vapour
will be richer in the more volatile component.
The aim is to balance the temperature of the column so that
by the time vapour reaches the top after huge numbers of condensing and reboiling operations, it consists only of the more
volatile component - in this case, B.
Whether or not this is possible depends on the difference
between the boiling points of the two liquids. The closer they are together, the longer the column has to be.
The liquid
So what about the liquid left behind at each reboiling? Obviously,
if the vapour is richer in the more volatile component, the liquid left behind must be getting richer in the other one.
As the condensed liquid trickles down the column constantly
being reboiled by up-coming vapour, each reboiling makes it richer and richer in the less volatile component - in this case,
A. By the time the liquid drips back into the flask, it will be very rich in A indeed.
So, over time, as B passes out of the top of the column into
the condenser, the liquid in the flask will become richer in A. If you are very, very careful over temperature control, eventually
you will have separated the mixture into B in the collecting flask and A in the original flask.
Finally, what is the point of the packing in the column?
To make the boiling-condensing-reboiling process as effective
as possible, it has to happen over and over again. By having a lot of surface area inside the column, you aim to have the
maximum possible contact between the liquid trickling down and the hot vapour rising.
If you didn't have the packing, the liquid would all be on
the sides of the condenser, while most of the vapour would be going up the middle and never come into contact with it.
Fractional distillation industrially
There is no difference whatsoever in the theory involved.
All that is different is what the fractionating column looks like. The diagram shows a simplified cross-section through a
small part of a typical column.
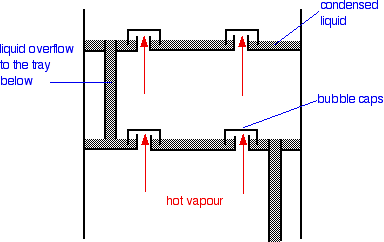
The column contains a number of trays that the liquid collects
on as the vapour condenses. The up-coming hot vapour is forced through the liquid on the trays by passing through a number
of bubble caps. This produces the maximum possible contact between the vapour and liquid. This all makes the boiling-condensing-reboiling
process as efficient as possible.
The overflow pipes are simply a controlled way of letting
liquid trickle down the column.
SOURCE:http://www.chemguide.co.uk/index.html#top